
ΑΙhub.org
An iterative refinement model for PROTAC-induced structure prediction
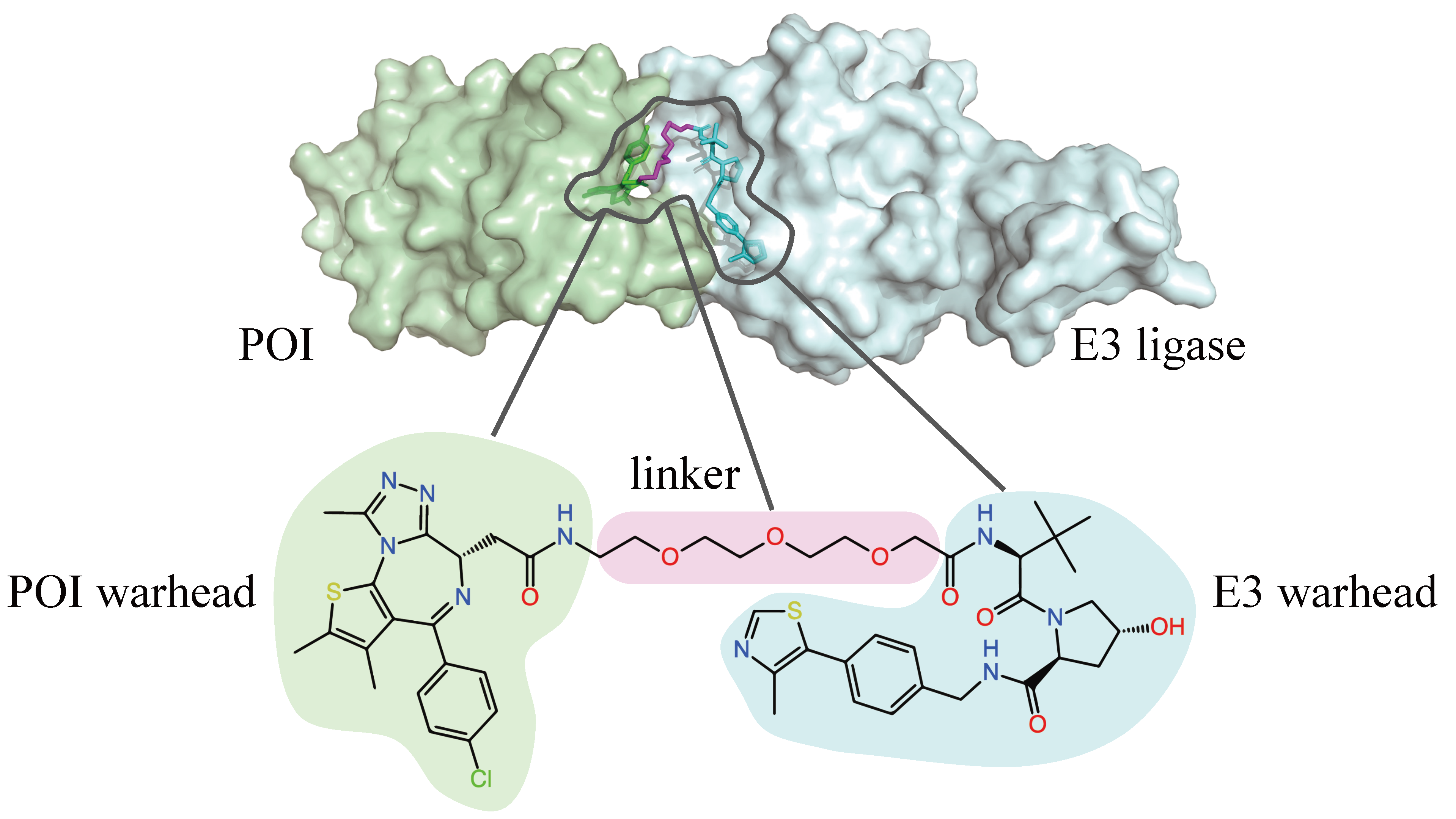
This work was accepted as an oral presentation at the Generative and Experimental Perspectives for Biomolecular Design workshop at ICLR 2024. For more information, please check out our paper on arXiv.
What are PROTACs?
Proteins are molecular machines that carry out many of the functions required for the human body to thrive. When proteins malfunction or over-accumulate, diseases may arise. Traditional small molecule drugs are designed to inhibit these disease-causing proteins by binding to them like keys (drug) fitting into locks (protein). However, binding does not always lead to therapeutic effect: only 27% of protein targets with known binders have an approved drug [1]. Proteolysis targeting chimeras, or PROTACs, are next-generation drug modalities designed to close this gap [2].
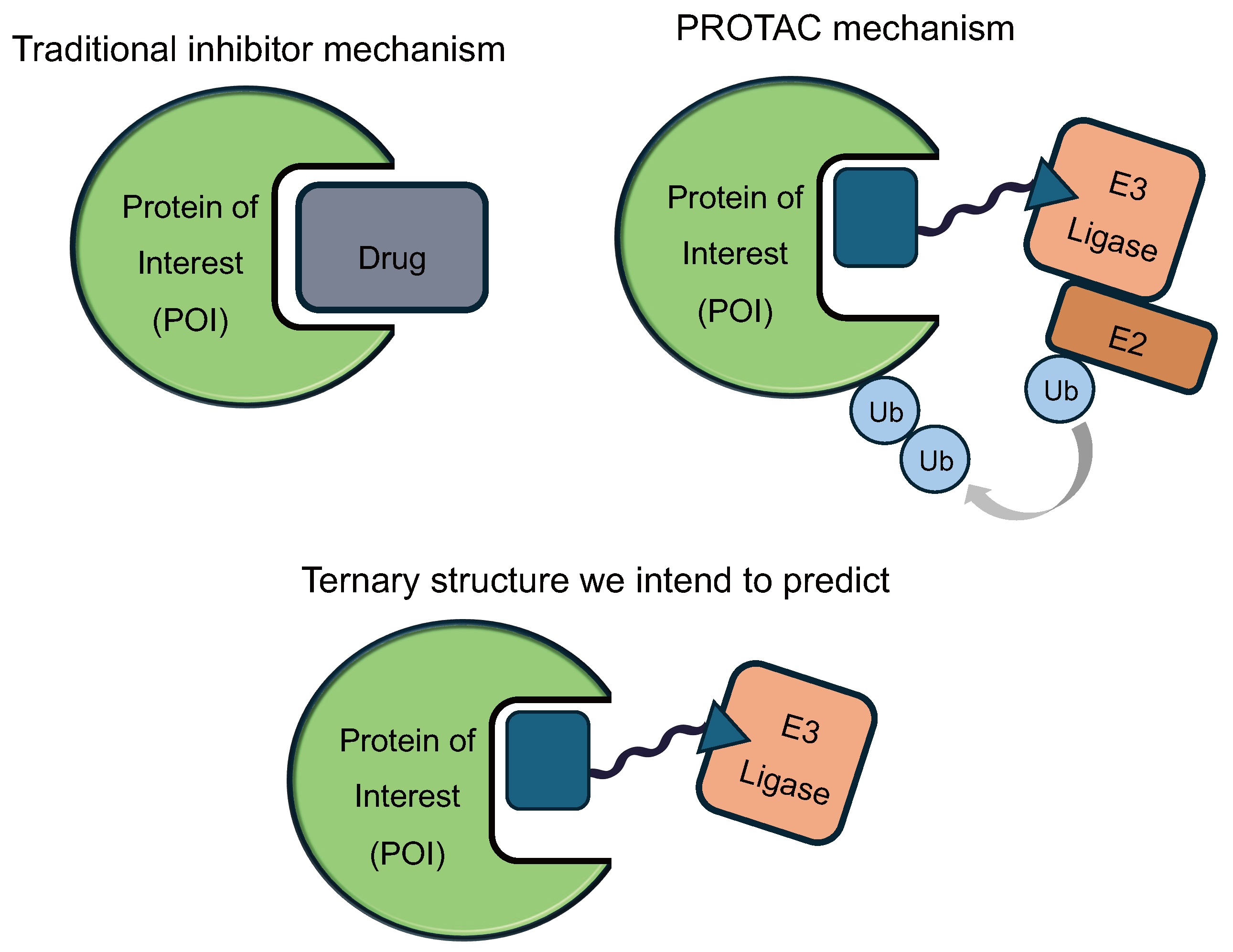
A PROTAC molecule can bind to both the target protein of interest (POI) and a another protein, known as an E3 ligase. The binding event brings the two proteins into proximity, which allows the E3 ligase to mark the POI for degradation. The tagged protein is then recognized by the proteasome, the cell’s garbage collection system, which breaks it down into non-functional pieces.
In this work, we aim to predict the ternary (3-way) structure of POI-PROTAC-E3, whose stability is critical to degradation activity. Understanding these structures will allow us to (1) choose the optimal E3 ligase for each protein target, (2) design the PROTAC linker to encourage stability, and (3) select the appropriate binders to facilitate this interaction.
Unfortunately, modeling these ternary structures has eluded both experimentalists and computationalists to this day. There are only 18 such structures in the PDB (Protein Data Bank), and PROTAC docking algorithms have yet to see widespread use, as existing search-based methods either are too slow or oversimplify the task.
Finally, deep learning has shown substantial promise in predicting the structures between a molecule and a single protein [3], or between pairs of proteins [4]. However, there are currently no end-to-end deep learning methods for PROTACs, due to the lack of data.
The PROFLOW framework
Creating realistic training data
To train a deep learning model in the absence of real ternary structures, we created a pseudo-ternary dataset by pairing binary protein-protein data with appropriate PROTAC linkers.
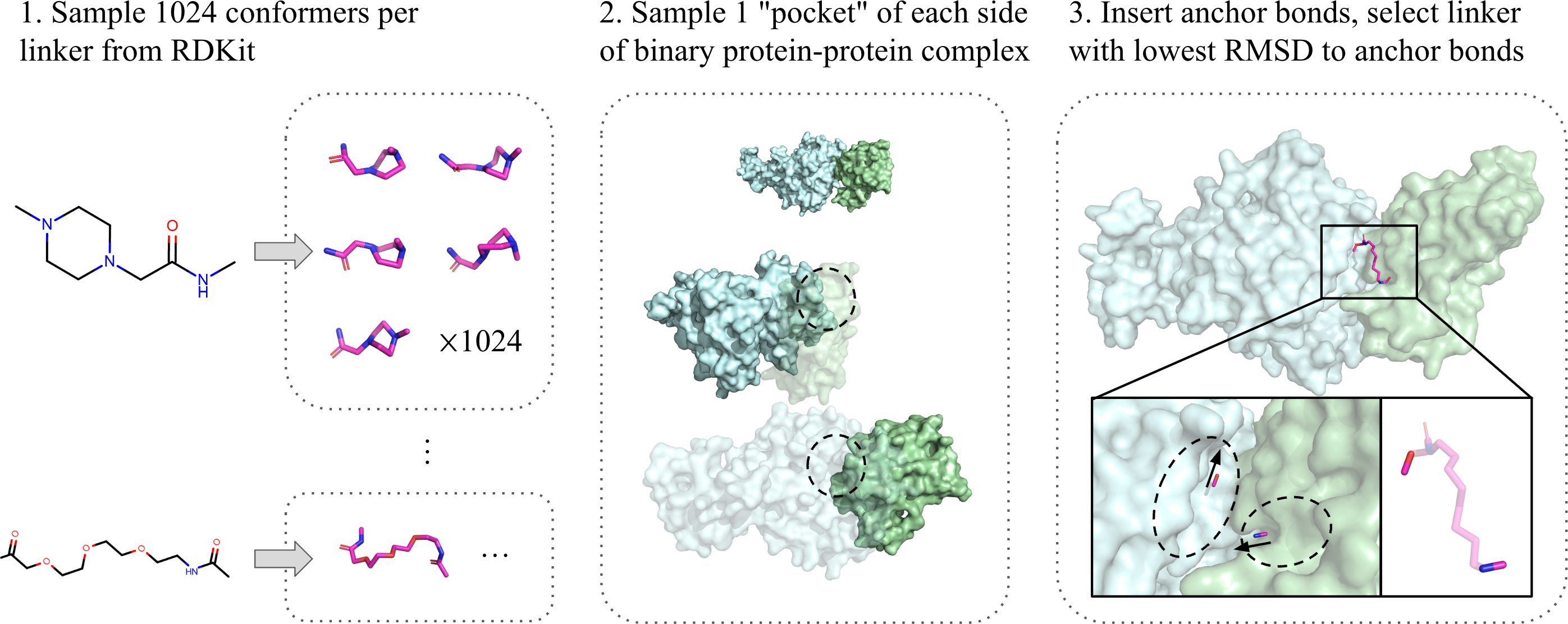
First, we generated a “linker library” by sampling 1024 conformations (chemically-plausible 3D coordinates) of each of the 1507 linkers reported in PROTAC-DB [5]. Next, for each of the 42k protein-protein pairs [6], we identified several “pocket” candidates on each protein, based on high-curvature patches on their surfaces. Finally, we assigned linker conformations to each pair of pockets by selecting the one whose geometry matches the closest.
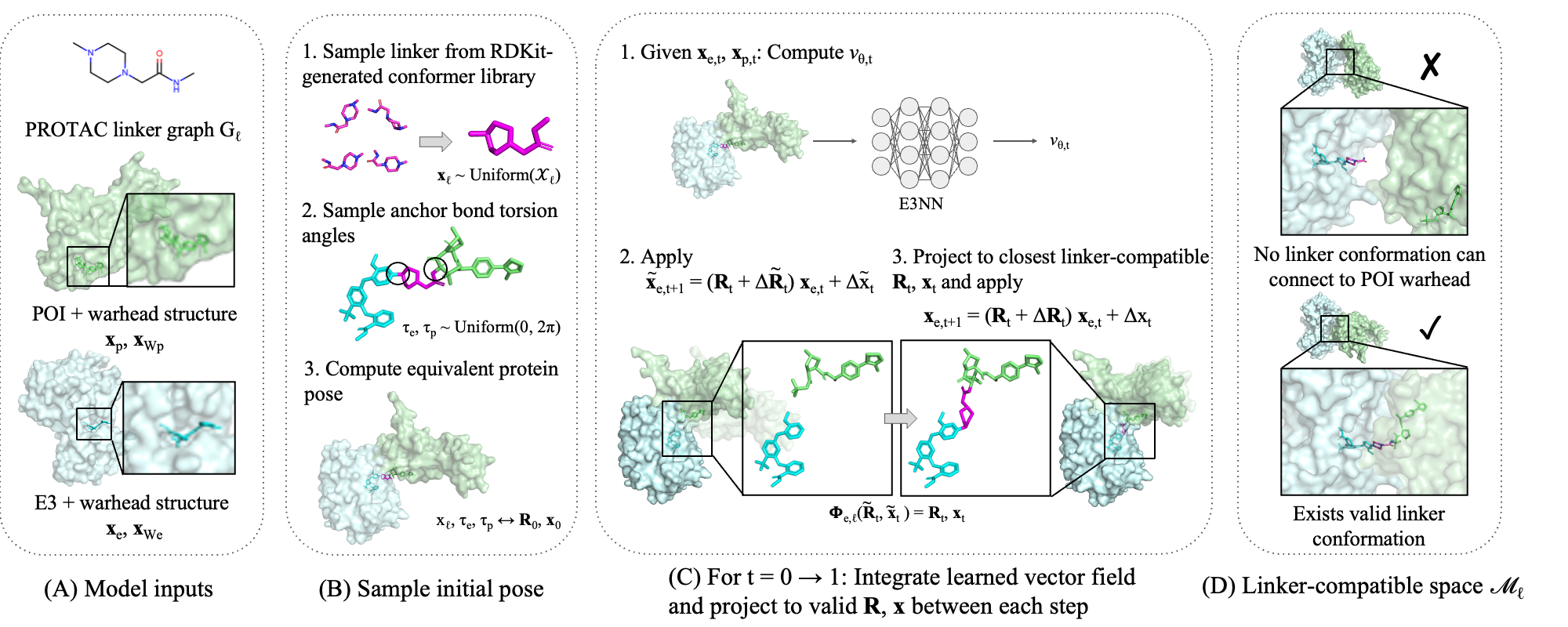
Generative model for ternary structures
We predict ternary structures using an iterative refinement framework inspired by flow matching [7], originally developed as generative AI for images. On a high level, our model starts from a random orientation of the two proteins, and iteratively updates the structures towards the “correct” conformations, as defined by ground truth. Specifically, at each step, the model takes as input the structures of the two proteins, and outputs the direction in which their relative rotation and translation should be updated. However, if we randomly manipulate the two proteins relative to one another, we cannot guarantee that the PROTAC can actually connect the two, as molecules cannot “stretch” arbitrarily. Therefore, before taking each step, we map the model’s proposed update to the closest update that can be achieved by the PROTAC (in terms of its set of linker conformers). To obtain the final ternary structure, we run the model for many steps, projecting back to the linker-compatible space each time.
Key results
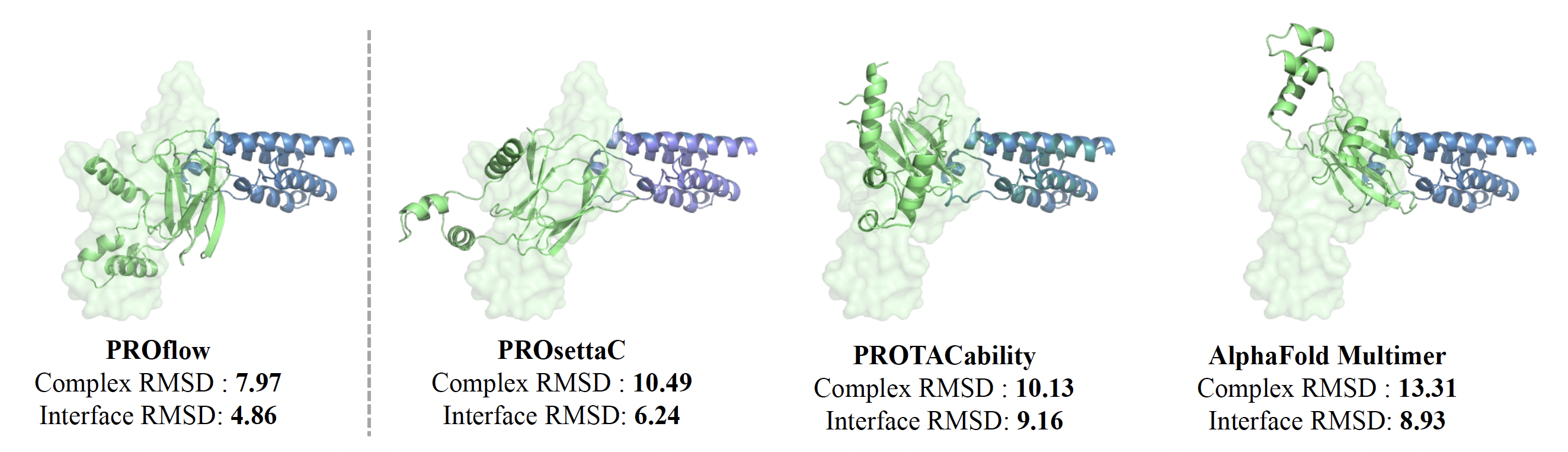
We conducted a comprehensive evaluation of PROTAC docking models, including several baselines like Alphafold-Multimer [4], PROsettaC [8] and PROTACability [9]. We assessed these models using the limited number of experimentally determined structures. In particular, we benchmarked their ability to recreate the E3-POI structures, using as input, either their individual structures, as extracted from the ground truth (holo), or from structures of the same proteins from independent experiments (apo).
When using bound (holo) structures, our model outperformed all baselines in structure prediction metrics like interface RMSD and Fnat, and matched the best baseline in complex RMSD. Baselines that ignored the PROTAC (Alphafold-Multimer) performed the worst in complex RMSD. In addition, PROFLOW achieved the fastest runtime, averaging 44 seconds per complex. When evaluated on unbound (apo) structures, PROFLOW maintained high performance, while other baselines showed a significant drop, highlighting their reliance on holo conformations.
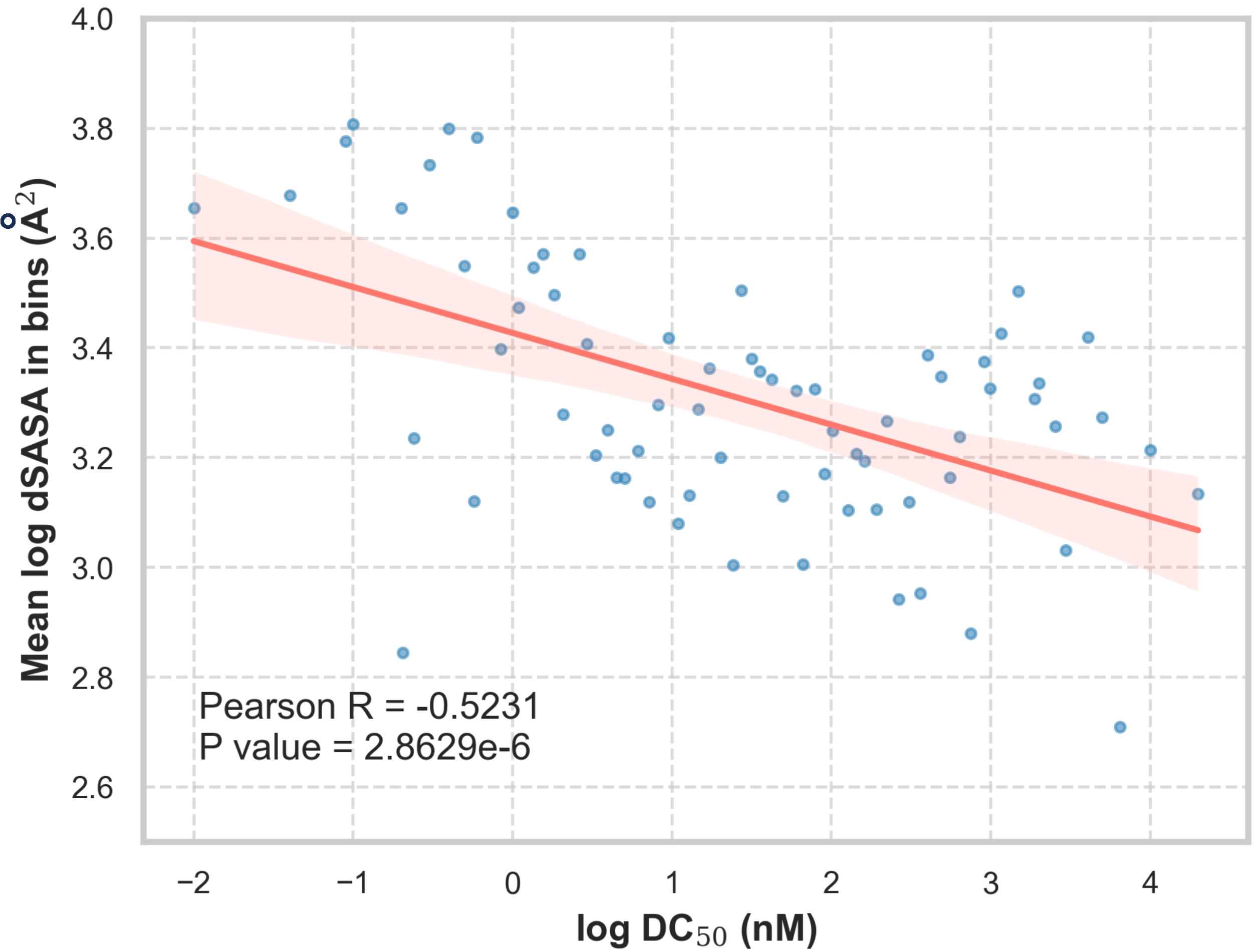
Since PROFLOW is orders of magnitude faster than alternatives, we were able to screen PROTAC designs at scale. We collected 483 examples with annotated DC50 values (the concentration of PROTAC required to degrade 50% of the POI), whose unbound E3 and POI structures were available in the PDB.
We observed a statistically significant correlation between solvent-accessible area buried at the interface (dSASA) and DC50 (p-value<0.001) with a Pearson correlation coefficient of -0.5231. The direction of this correlation is biologically intuitive: PROTACs that induce larger interfaces are more potent degraders.
In summary, our iterative refinement model, PROFLOW, offers a novel approach to predicting PROTAC-induced ternary structures by leveraging a pseudo-ternary dataset. PROFLOW outperforms existing models in both accuracy and runtime, and shows correlation of predicted interfaces area and degradation activity. Therefore, we hope that this work will facilitate the understanding and rational design of PROTACs in the future.
References
[1] https://pharos.nih.gov/targets
[2] Jonathan M. Tsai et al, Targeted protein degradation: from mechanisms to clinic, Nature Reviews Molecular Cell Biology, 2024
[3] Gabriele Corso et al, DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking, International Conference on Learning Representations (ICLR 2023)
[4] Richard Evans et al, Protein complex prediction with AlphaFold-Multimer, Biorxiv, 2022
[5] Gaoqi Weng et al, PROTAC-DB 2.0: an updated database of PROTACs, Nucleic Acids Research, 2022
[6] Alex Morehead et al, DIPS-Plus: The enhanced database of interacting protein structures for interface prediction, Scientific Data, 2023
[7] Yaron Lipman et al, Flow Matching for Generative Modeling, International Conference on Learning Representations (ICLR 2023)
[8] Daniel Zaidman et al, PRosettaC: Rosetta Based Modeling of PROTAC Mediated Ternary Complexes, Journal of Chemical Information and Modeling, 2020
[9] Gilberto P. Pereira et al, Rational Prediction of PROTAC-Compatible Protein–Protein Interfaces by Molecular Docking, Journal of Chemical Information and Modeling, 2023
Acknowledgements
We thank Hannes Stark, Jason Yim, Bowen Jing, and Gabriele Corso for helpful discussions.
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1745302. We would like to acknowledge support from the NSF Expeditions grant (award 1918839: Collaborative Research: Understanding the World Through Code), Machine Learning for Pharmaceutical Discovery and Synthesis (MLPDS) consortium, the Abdul Latif Jameel Clinic for Machine Learning in Health, and the DTRA Discovery of Medical Countermeasures Against New and Emerging (DOMANE) threats program.
tags: ICLR2024




AIhub is supported by:








