
ΑΙhub.org
Using machine learning to improve the toxicity assessment of chemicals
Researchers from the University of Amsterdam, together with colleagues at the University of Queensland and the Norwegian Institute for Water Research, have developed a strategy for assessing the toxicity of chemicals using machine learning. They present their approach in an article in Environmental Science & Technology. The models developed in this study can lead to substantial improvements when compared to conventional ‘in silico’ assessments based on quantitative structure-activity relationship (QSAR) modelling.
According to the researchers, the use of machine learning can vastly improve the hazard assessment of molecules, both in the safe-by-design development of new chemicals and in the evaluation of existing chemicals. The importance of the latter is illustrated by the fact that European and US chemical agencies have listed approximately 800,000 chemicals that have been developed over the years but for which there is little to no knowledge about environmental fate or toxicity.
Since an experimental assessment of chemical fate and toxicity requires much time, effort, and resources, modelling approaches are already used to predict hazard indicators. In particular quantitative structure-activity relationship (QSAR) modelling is often applied, relating molecular features such as atomic arrangement and 3D structure to physicochemical properties and biological activity. Based on the modelling results (or measured data where available), experts classify a molecule into categories as defined for example in the Globally Harmonized System of Classification and Labelling of Chemicals (GHS). For specific categories, molecules are then subjected to more research, more active monitoring and, eventually, legislation.
However, this process has inherent drawbacks, many of which can be traced back to the limitations of the QSAR models. They are often based on very homogeneous training sets and assume a linear structure-activity relationship for making extrapolations. As a result, many chemicals are not well-represented by existing QSAR models and their use can potentially lead to substantial prediction errors and misclassification of chemicals.
Skipping the QSAR prediction
In the paper published in Environmental Science & Technology, Dr Saer Samanipour and co-authors propose an alternative evaluation strategy that skips the QSAR prediction step altogether. Samanipour, an environmental analytical scientist at the University of Amsterdam’s Van’t Hoff Institute for Molecular Sciences teamed up with Dr Antonia Praetorius, an environmental chemist at the Institute for Biodiversity and Ecosystem Dynamics of the same university. Together with colleagues at the University of Queensland and the Norwegian Institute for Water Research, they developed a machine learning-based strategy for the direct classification of acute aquatic toxicity of chemicals based on molecular descriptors.
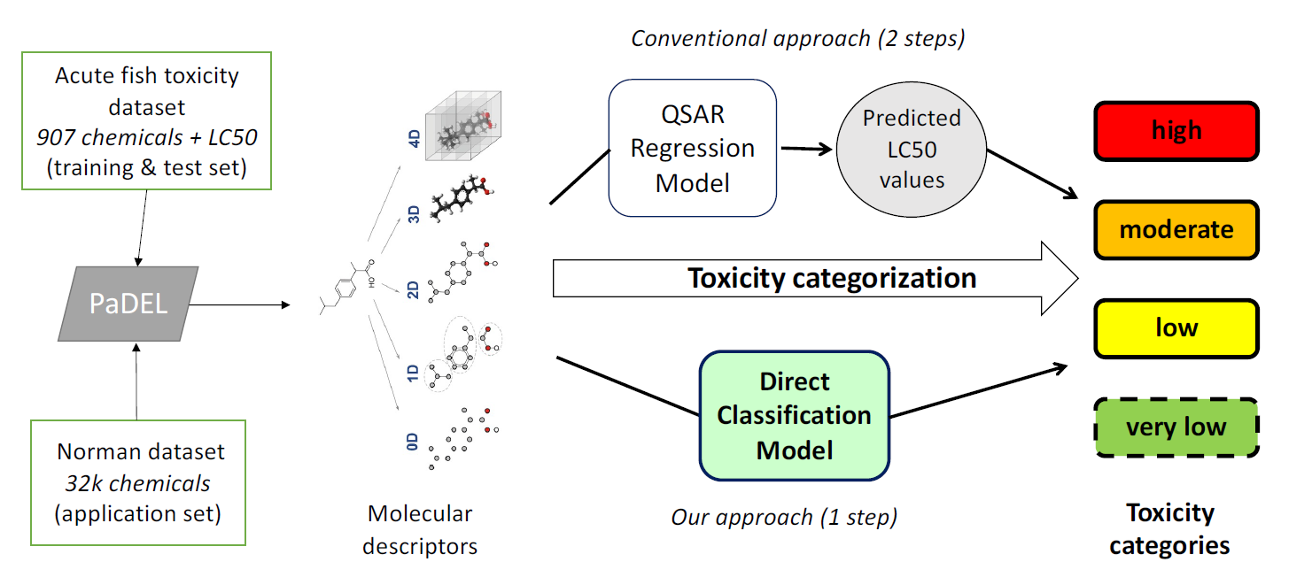 Overall workflow of the study, from the raw data to the finally generated models. Image taken from the article “From Molecular Descriptors to Intrinsic Fish Toxicity of Chemicals: An Alternative Approach to Chemical Prioritization”.
Overall workflow of the study, from the raw data to the finally generated models. Image taken from the article “From Molecular Descriptors to Intrinsic Fish Toxicity of Chemicals: An Alternative Approach to Chemical Prioritization”.
The model was developed and tested via 907 experimentally obtained data for acute fish toxicity (96h LC50 values). The new model skips the explicit prediction of a toxicity value (96h LC50) for each chemical, but directly classifies each chemical into a number of pre-defined toxicity categories. These categories can for example be defined by specific regulations or standardization systems, as demonstrated in the article with the GHS categories for acute aquatic hazard. The model explained around 90% of the variance in the data used in the training set and around 80% for the test set data.
Higher accuracy predictions
This direct classification strategy resulted in a fivefold decrease in the incorrect categorization compared to a strategy based on a QSAR regression model. Subsequently, the researchers expanded their strategy to predict the toxicity categories of a large set of 32,000 chemicals.
They demonstrate that their direct classification approach results in higher accuracy predictions because experimental datasets from different sources and for different chemical families can be grouped to generate larger training sets. It can be adapted to different predefined categories as prescribed by various international regulations and classification or labelling systems. In the future, the direct classification approach could also be expanded to other hazard categories (e.g. chronic toxicity) as well as to environmental fate (e.g. mobility or persistence) and shows great potential for improving in-silico tools for chemical hazard and risk assessment.
Paper details
From Molecular Descriptors to Intrinsic Fish Toxicity of Chemicals: An Alternative Approach to Chemical Prioritization, Saer Samanipour, Jake W. O’Brien, Malcolm J. Reid, Kevin V. Thomas, and Antonia Praetorius. Environ. Sci. Technol. 2022.
AUAI is supported by:








